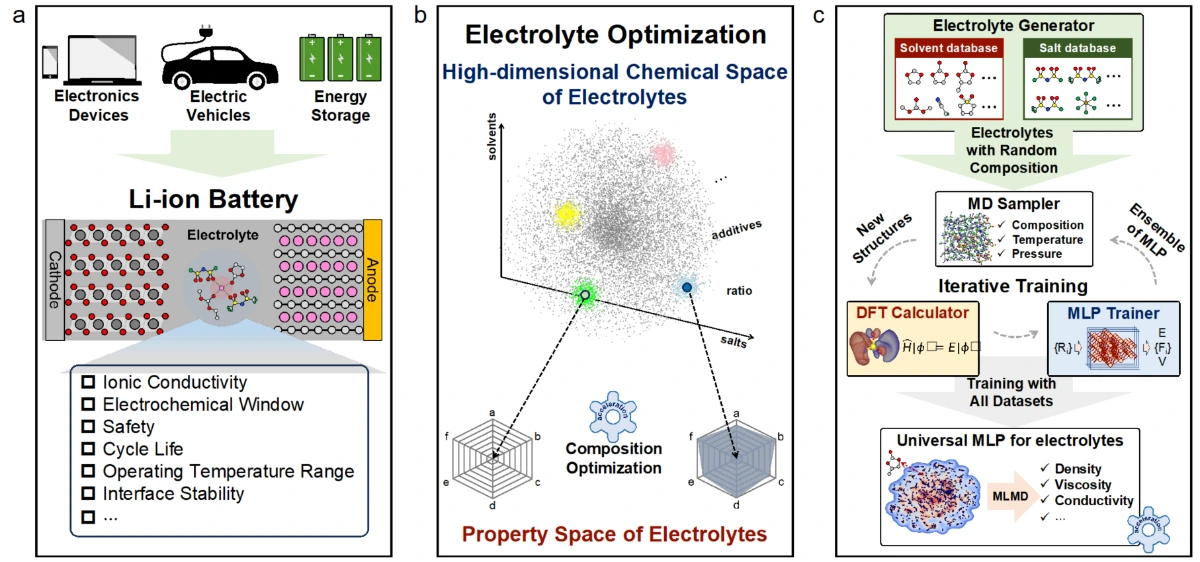
【导读】
锂离子电池广泛应用于电子设备、电动汽车和航空领域,对高能量密度、快充能力和宽工作温度范围有着极高要求。计算与实验相结合的方法在电解质开发中日益受到关注,然而,电解质固有的复杂性带来了巨大挑战。经典分子动力学(CMD)常因力场参数不准确而失效,而从头算计算(ab initio)则受限于高昂的计算成本。
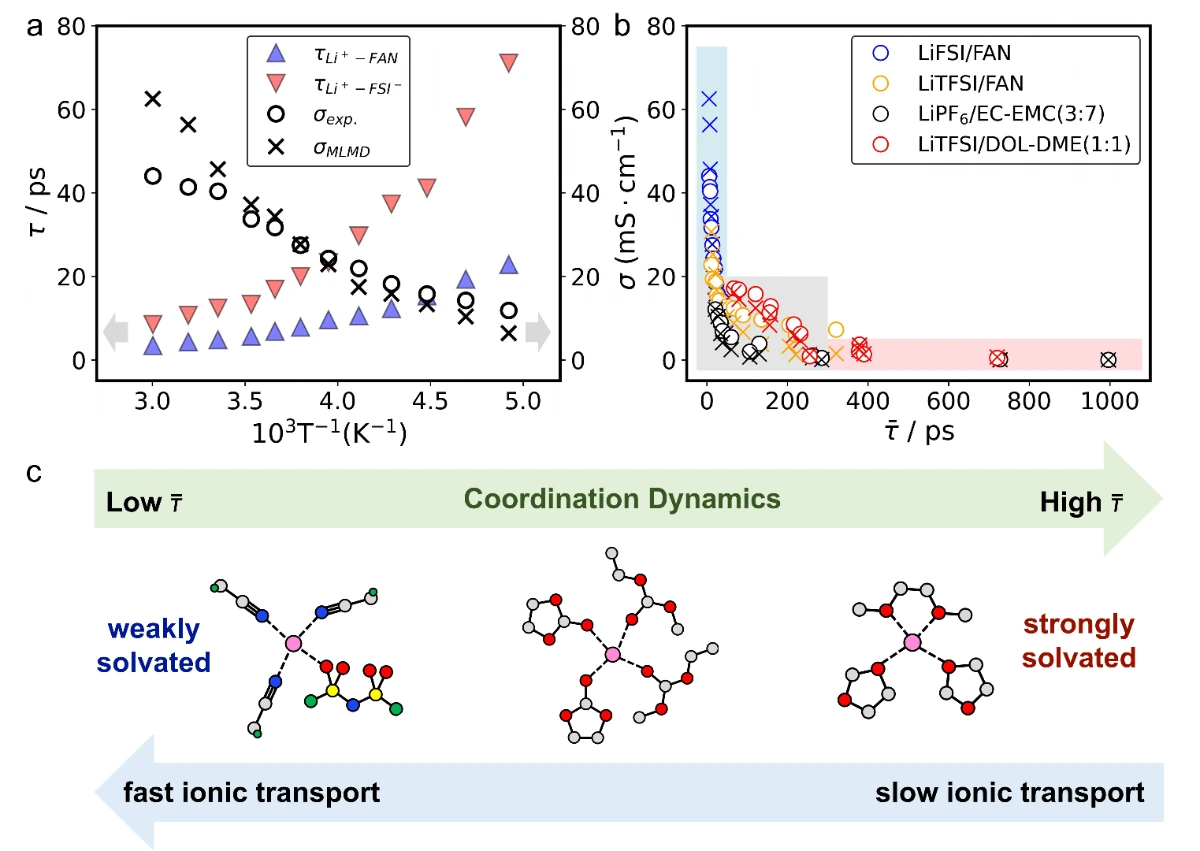
近年来,机器学习分子动力学(MLMD)作为一种高效且准确的替代方案脱颖而出,但其应用受限于机器学习势有限的泛化性(迁移性)。在这项工作中,我们通过对随机组合的数据集进行迭代训练,开发了一种面向电解质的通用机器学习势(uMLP),能够利用分子动力学准确计算各种电解质的关键性质。此外,通过量化配位寿命对锂离子进行的配位动力学分析,为溶剂化强度提供了一种直接且定量的衡量标准。
【成果掠影】
本工作利用随机电解液生成器和基于ai2-kit的并发学习工作流,构建了覆盖超2300种溶剂和20种锂盐的通用机器学习势函数(uMLP)。该势函数不仅具有从头算的精度,还能实现纳秒级的长时分子动力学模拟,有效克服了传统方法在的局限。在此基础上,团队利用该通用势函数系统地计算了多种电解液的密度、粘度、离子电导率及工作温域,计算结果与实验数据高度吻合。更重要的是,该工作通过配位动力学分析,提出了以配位寿命作为定量描述溶剂化强度的指标。研究发现,较短的配位寿命直接对应于“弱溶剂化”结构,有利于离子的快速传输。这一发现不仅揭示了微观溶剂化结构与宏观电化学性能之间的内在联系,也为设计高性能的电解液提供了理论指导。
【数据】
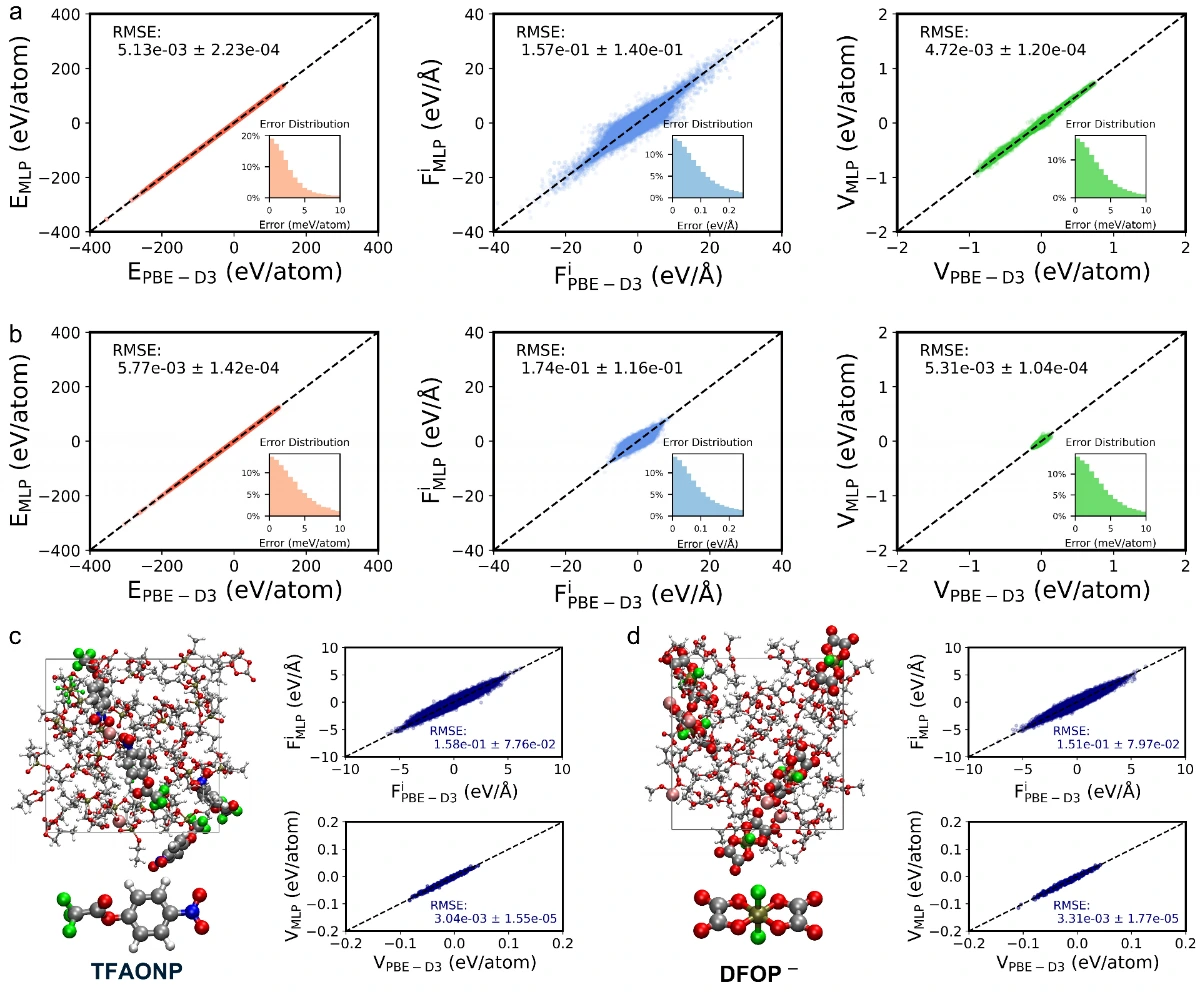
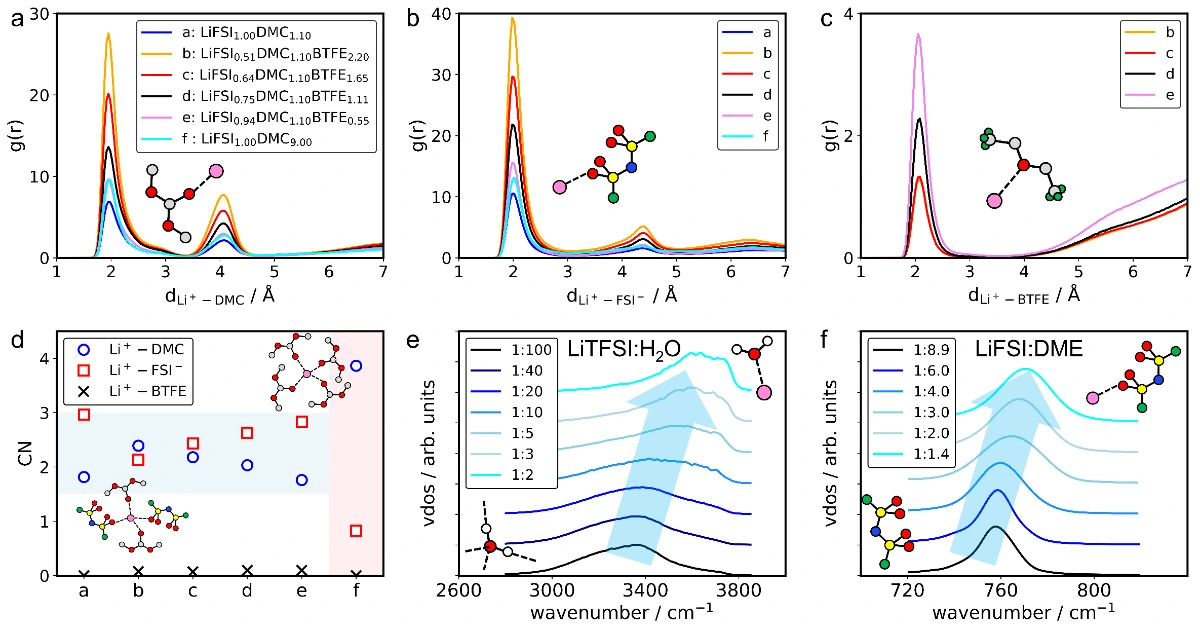
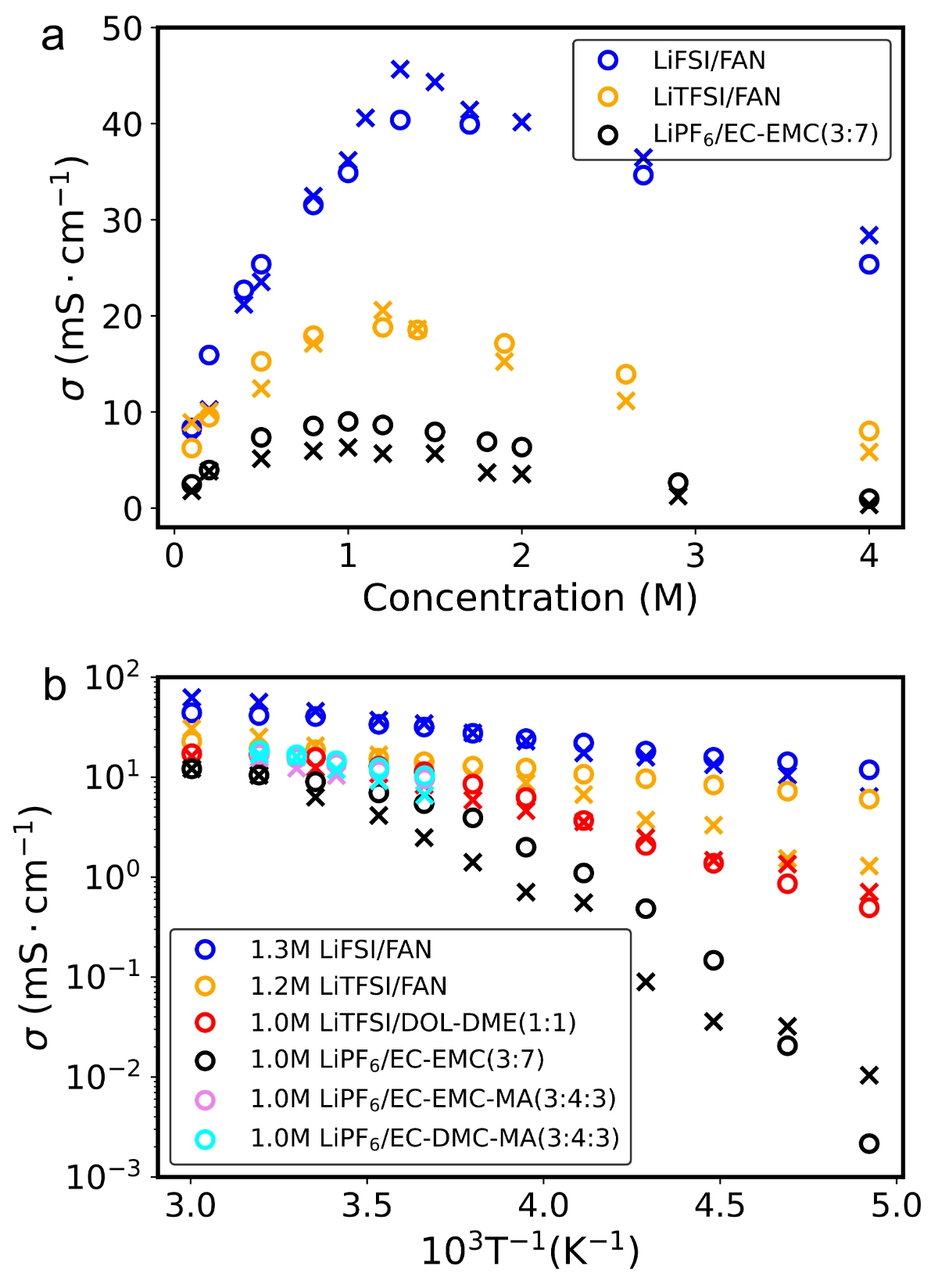
【致谢】
该工作在程俊教授指导下,嘉庚创新实验室王锋副研究员和博士生唐煜航为共同第一作者,博士生马泽冰、金昱丞参与了该项工作。该论文得到了国家自然科学基金(22225302, 92470201, 92461312, 22021001, 21991151, 21991150, 92161113, 22411560277, 22393901, 22503037)、中央高校基本科研业务费(20720250005, 20720220009, 20720230090)以及嘉庚创新实验室和人工智能应用电化学联合实验室(RD2023100101, RD2022070501)的资助。
程俊教授课题组长期致力于将机器学习方法应用于复杂能源材料的模拟与设计。本工作进一步展示了通用机器学习势函数在复杂液态体系中的应用潜力。本工作相关的数据集和势函数模型已在AI4EC仓库开源(https://doi.org/10.12463/AI4EC/QZCYP1),并在 op-elyte emulator (https://ai4ec.ac.cn/apps/op-elyte-emulator) 提供在线试用。
