研究背景
过去二十年间,以“吸附能”为核心的描述符理论主导了电催化领域。通过评估反应中间体的热力学稳定性,该框架成功解释了大量实验现象并指导了催化剂的理性设计,至今仍是该领域不可动摇的理论基石。
然而,“吸附能”本质上是一个宏观的热力学量。它能精准地告诉我们反应“难易如何”,但在揭示“为什么难易”这一微观电子结构层面的本征物理过程时,往往存在视角上的盲区。
面对这一挑战,本工作选择将视角下沉至凝聚态物理的底层逻辑,试图从电子态杂化的本源去理解电催化过程。
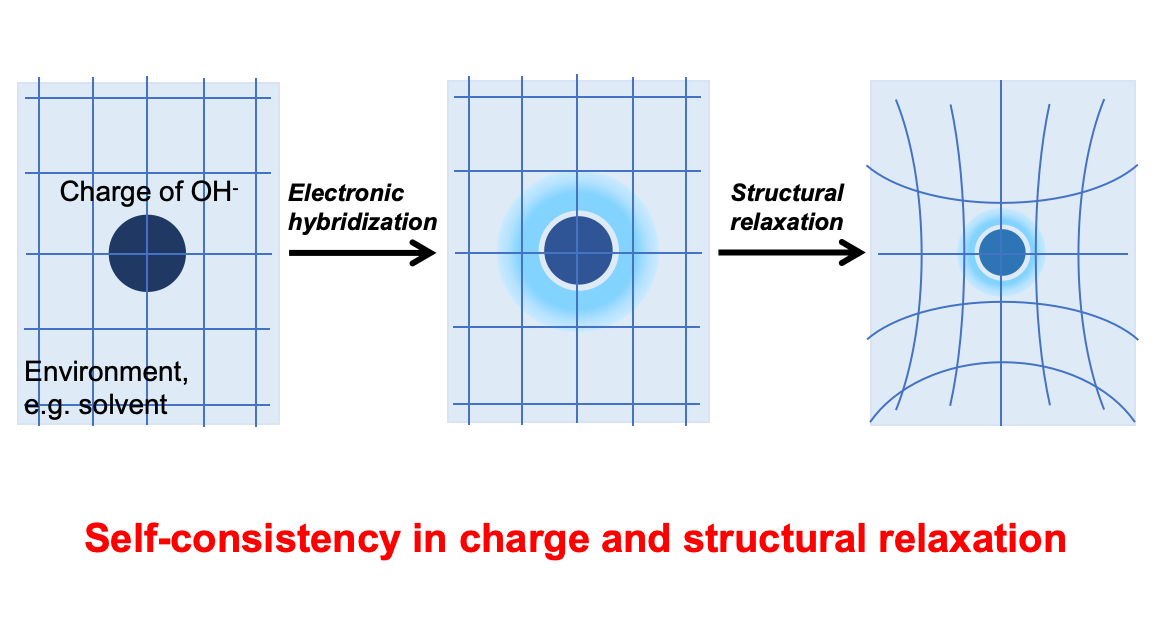
核心理论突破
基于基本的物理化学原理,半导体的催化效应是通过能带结构与反应离子的杂化实现的。能带的杂化会使得反应离子的部分电荷离域进入能带,从而使得离子的氧化态和还原态偏离离子近似。这种电荷的离域会影响溶剂的响应,而溶剂响应的变化又会反过来影响反应离子与能带结构的杂化。可以看到,“能带杂化”、“氧化态/还原态的电荷离域”、“溶剂效应”相互之间的这种耦合,意味着我们需要一个闭环的自洽理论来进行描述。
几十年来,尽管基于Anderson-Newns哈密顿量的电化学理论(如经典的Schmickler模型)极大地推动了我们对电子转移过程的理解,然而,上述的全自洽理论仍然是缺失的。为此,在本工作中,研究团队另辟蹊径,将半导体缺陷物理中用于描述价态变化时电荷离域效应的 Haldane-Anderson 格林函数理论,与描述溶剂效应的 Holstein 模型相结合,成功地在一个统一的格林函数框架下,实现了对“能带杂化-电荷离域-溶剂效应”的全自洽处理。
全新视角带来的物理洞见
基于这一全自洽模型,本工作为几个长期悬而未决的关键科学问题提供了底层的物理归因:
1. 阐明半导体电催化活性的微观电子起源 自洽模型计算表明,半导体能带杂化降低反应活化能的根本原因在于:在自洽框架下,氧化还原过程中实际转移的局域电荷量显著减少。转移电荷的减少直接削弱了溶剂重组的幅度(重组能降低),从而降低了反应能垒。这为半导体催化剂的活性来源提供了一个超越宏观吸附能的微观电子结构解释。
2. 揭示重组能不对称性的电子结构起源 在AIMD模拟中,常观察到氧化与还原过程的重组能不相等,传统观点难以给出本源解释。我们的全自洽模型证明:即使假设溶剂是线性响应,只要存在能带杂化,重组能的不对称性就是物理上的必然结果。其根源在于,在自洽框架下,氧化和还原过程中真正发生转移的“束缚态电荷”是不同的。这一发现从模型层面澄清了该现象的物理起源。
总结与展望
在算力飞速发展的今天,DFT等第一性原理计算已成为研究催化机制的标准工具,但其“黑箱”特性有时会掩盖底层的物理直觉。
本工作表明,模型哈密顿量方法并未过时。虽然它在定量精度上不及DFT计算,但本文构建的全自洽模型在提供“白箱”式的物理图像方面具有不可替代的优势。通过将“氧化还原反应”与“缺陷电荷态转变”建立起同构映射,我们不仅为半导体电催化提供了一个坚实的理论补充,也为未来在更复杂的电极/溶液界面体系中理解电子转移行为开辟了一条基于凝聚态物理的新途径。
