导读
电化学界面在能源存储与转化中至关重要,物种、结构复杂而且发生动态变化,需要采用实验表征和理论计算共同进行研究。但是,现有可实现对界面准确描述的理论计算方法(如第一性原理分子动力学)极为昂贵,使得研究局限在百原子、十皮秒量级的模型体系,且界面的原位环境和动态特征只能分离模拟。然而,实际电化学体系更加复杂,需同时考虑原位和动态,在更大的时间和空间尺度进行模拟。近年来快速发展的机器学习势函数能够实现在保持第一性原理精度的前提下大幅加速计算,为实现对原位、动态的复杂电化学界面的模拟提供了技术基础。然而,将机器学习势函数应用于电化学界面模拟仍存在诸多挑战。一方面,介电响应等性质需要对长程静电相互作用进行准确描述;另一方面,金属电极(电子导体、离子绝缘体)和电解液(离子导体、电子绝缘体)的介电响应性质差异显著,现有机器学习模型难以同时描述。
研究内容
在电化学界面处,可能存在的高电场意味着机器学习势函数需要描述界面极化随电场的变化行为(即界面介电响应)。在介电理论中,电介质对电场的响应方式主要包括:离子定向迁移、偶极子的旋转、分子内部化学键的形变以及电子极化。对于常见的电化学界面,电解质为离子导体和电子绝缘体,在零电荷电势附近介电响应主要由前三种机制主导,而电子重排(即电子介电响应)的贡献相对较小。以体相水为例,其在常温常压下的介电常数约为78,而电子介电响应对应的贡献仅约为1.8。与之相反,金属电极是电子导体和离子绝缘体,其介电响应主要通过电子极化实现。电解质和电极不同的介电响应导致在ec-MLP中描述其响应时需要采用不同的方法:电解质采用基于Wannier质心的方法,而电极采用Siepmann-Sprik可极化电极方法。
对于离子导体和电子绝缘体的电解质中,介电响应主要由原子和离子的移动承担。因此,我们选择基于Wannier质心的方法来描述电解质介电响应。在该方法中,基于局域化学环境预测Wannier质心相对于对应原子核的位置。与电解质不同,在金属电极中,原子的位移通常可以忽略,而电子极化产生的介电响应(即电子介电响应)起重要作用。此前,在经典分子动力学模拟领域已经存在一些方法来描述金属的电子介电响应。例如,在Siepmann-Sprik可极化电极方法中,电极中的电荷分布通过位于中心位于原子坐标的球形高斯分布来近似。在此方法中,体系能量为原子电量的二次函数,可通过能量极小化求得在特定外场和环境(如电解质构型)下的电荷分布。
基于上述分析,我们将基于Wannier质心的方法和可极化电极方法联用,用于表示金属-电解质界面的介电响应(如图1所示)。对于给定的原子构型,Wannier质心的位置通过一个用于预测原子张量性质的机器学习模型来确定,而电解质中的电荷分布被近似为位于原子核和Wannier质心位置的点电荷。基于这种电荷分布,可以计算电解质在电极原子位置产生的静电势,进而可以在给定静电边界条件下求解电极原子的电荷分布。界面处的电荷分布(包括电解质和电极)可用于计算总能量中的长程静电贡献,而剩余的短程能量则可以通过基于局域化学环境的机器学习模型预测。在本工作中,我们选择Deep Wannier深度神经网络模型来描述电解质的介电响应,而选择Siepmann-Sprik模型来描述金属电极的介电响应。
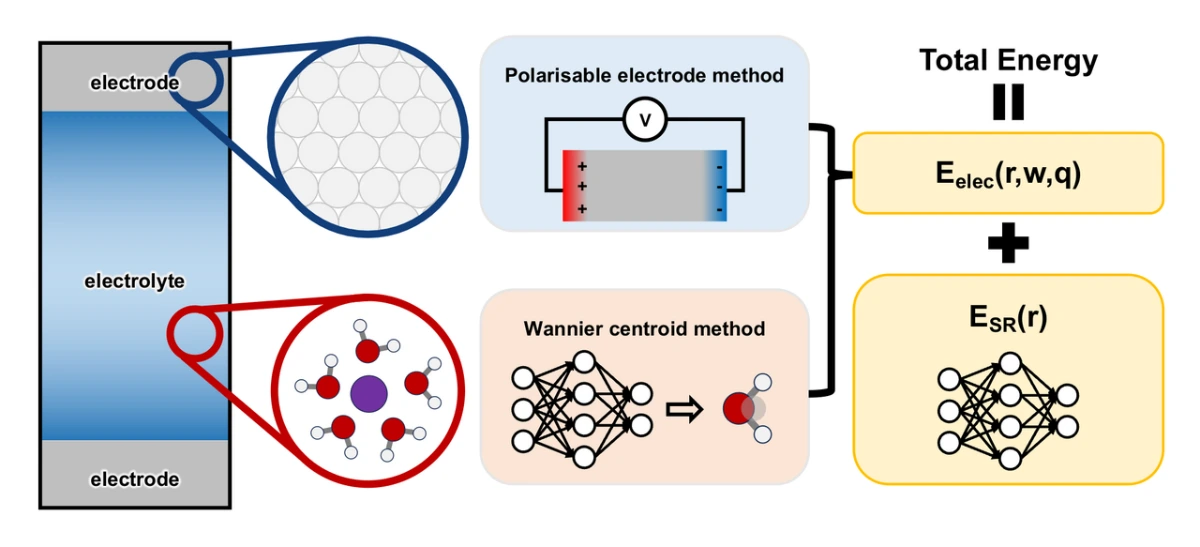
为了验证上述提出的应用于电化学界面模拟的机器学习势函数ec-MLP的有效性,我们选择Pt(111)-KF溶液界面作为模型体系。这一选择不仅因为Pt(111)-电解质界面在实验和计算电化学中被广泛用作模型体系,更重要的是,文献中表明,Pt表面由于施加电势引起的水分子动态化学吸附,表现出钟形的亥姆霍兹微分电容曲线。这一现象突出了同时描述电子结构和分子动力学效应的重要性,而这无法通过静态第一性原理计算或经典分子动力学模拟来全面描述。基于与前期第一性原理分子动力学模拟类似的计算设置,研究者成功描述Pt(111)-电解质界面的水随电极电势变化造成的化学吸附/脱附现象(见图2a和2b)及由此产生的钟形微分电容曲线(见图2c),证明了ec-MLP方法的有效性。
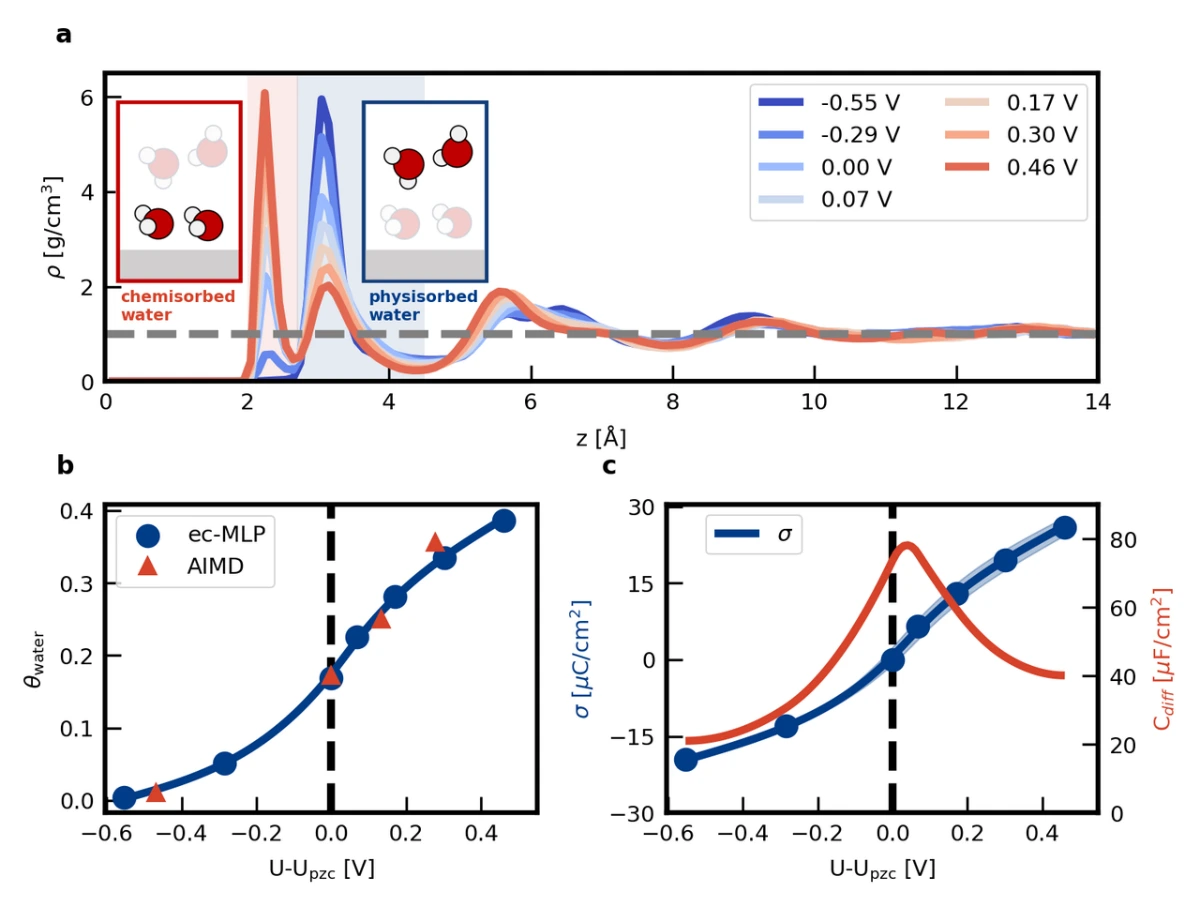
与体相水相比,在固体表面的水表现出独特的介电行为,这在实验和模拟中都引起了极大的兴趣。然而,我们对界面处介电常数如何变化的理解在定性层面上仍然不足。近些年来,分子动力学模拟使得获取沿垂直于表面方向的介电常数分布成为可能。然而,现有的介电分布研究仅限于惰性界面,对于高活性界面(如Pt-水界面)的计算仍然相对缺乏,这可能是由于以下原因:一方面,因为忽略了电子结构,经典力场模拟无法正确描述界面水结构;另一方面,当前密度泛函理论计算中,对包含导体组分的体系施加电场非常困难。
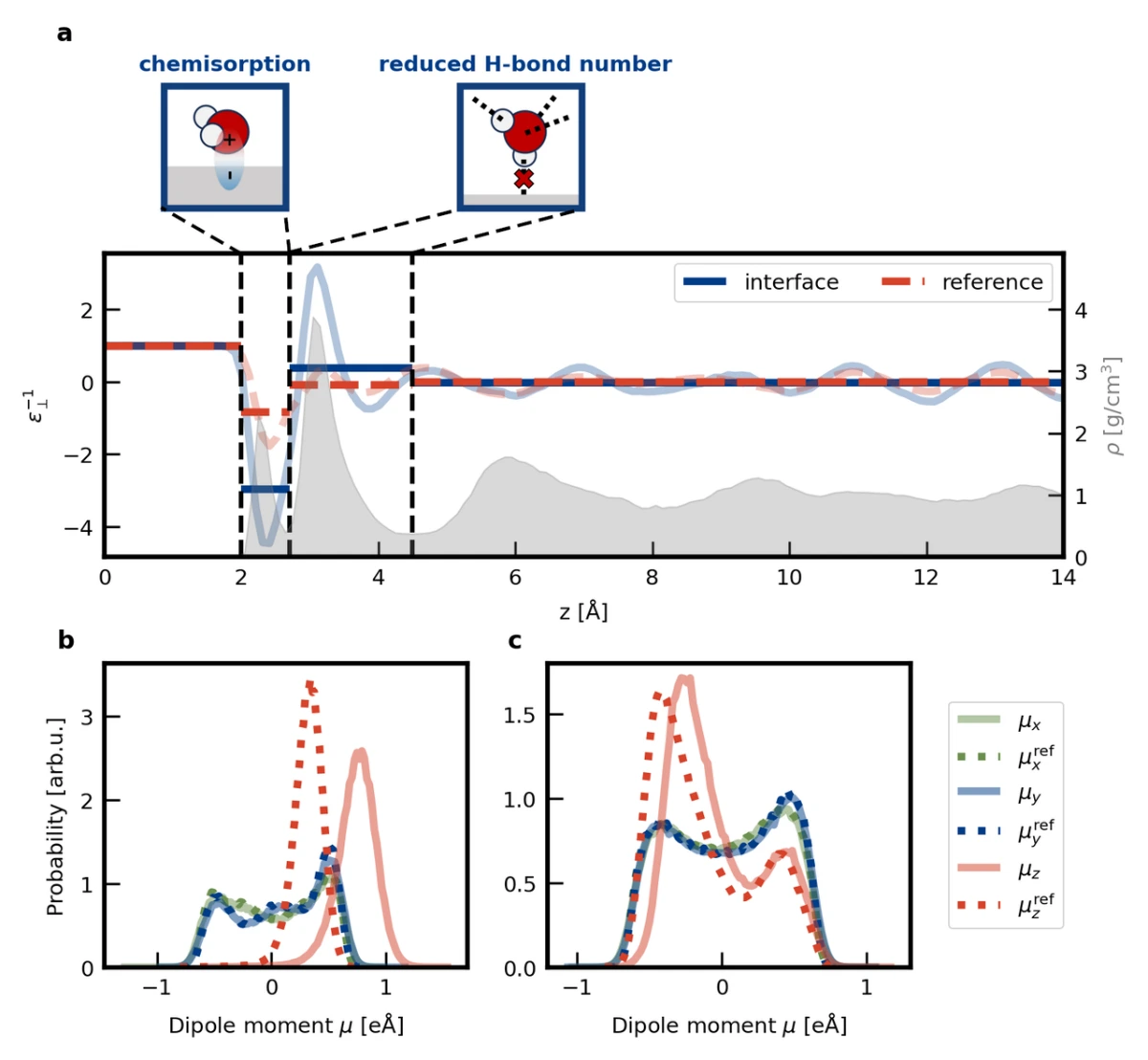
在本工作中,我们借助ec-MLP,首次计算了Pt(111)-水界面的介电常数空间分布(如图3a)。其中,化学吸附水区域(距离金属表面2.7埃以内)存在对电场的过屏蔽,而物理吸附水区域(距离金属表面2.7-4.5埃)的平均介电常数远小于体相水。对特定区域内水分子的偶极进行分析可知,化学吸附水区域的电场过屏蔽源于化学吸附诱导的分子偶极增大,而物理吸附水区域的介电常数偏小源于氢键数目降低。值得注意的是,前期工作通常认为非化学吸附水的介电常数降低源于转动受限,而本工作揭示了在非化学吸附水中电子极化也会对介电响应存在显著影响。
致谢
研究团队感谢乐家波博士、杨晓晖博士、Katharina Doblhoff-Dier博士、黄俊博士、张林峰博士、王涵博士在课题推进和文章写作方面的深入讨论和宝贵建议,以及深度势能社区以及开发团队成员徐伟鸿等提供的应用开发支持。朱嘉欣博士感谢厦门大学及2011能源材料化学协同创新中心提供的支持。程俊教授感谢中国国家自然科学基金(资助号:22225302, 92461312, 22021001, 21991151, 21991150, 92161113, 22411560277)、中央高校基础研究基金(资助号:20720220008, 20720220009, 20720220010)、人工智能应用电化学实验室(AI4EC)、IKKEM(资助号:RD2023100101 和 RD2022070501)的支持。
原文详情
https://journals.aps.org/prl/abstract/10.1103/48ct-3jxm
代码链接
ec-MLP相关功能近期将以功能模块形式发布于ai2-kit:
