研究背景:
固液界面对许多物理和化学过程都至关重要,和频光谱(SFG)作为一类特殊的界面敏感光谱学手段,已经用于对固液界面的广泛研究。要在特定光谱信号和独特的界面水结构之间建立明确的相关性,需要使用分子动力学(MD)模拟进行理论计算。这些模拟通常需要相对较长的轨迹(几纳秒),才能通过偶极矩-极化率时间相关函数实现可靠的SFG光谱计算。然而,对长轨迹的要求限制了成本高昂的技术如从头算分子动力学(AIMD)模拟的使用。在这项工作中提出了一种利用机器学习(ML)加速方法计算固液界面振动光谱(红外、拉曼、SFG 光谱)的途径。采用偶极矩-极化率关联函数(μ-α)和表面敏感速度-速度关联函数(ssVVCF)方法来计算 SFG 光谱。结果表明使用 ML 方法成功加速了 AIMD 模拟和 SFG 光谱计算。这一工作为借助 ML 方法以更低的计算成本更快地计算复杂固液界面的 SFG 光谱提供了机会,从而揭示电化学研究中至关重要的界面双电层提供了可能性。
理论与计算方法:
单点能计算:CP2K+PBE+D3
机器学习:Deep Potential(DP)模型和Deep Wannier(DW)模型
结果讨论与文献解析:
本文训练了Deep Potential(DP)模型和Deep Wannier(DW)模型,分别对分子结构的势能、力、偶极矩和极化率进行了预测。然后通过这些预测结果,快速且低成本地计算了α-Al2O3(0001)-水界面的各种振动光谱,且轨迹结构分析和振动光谱计算都表明ML模型达到了第一性原理的计算精度。
图1展示了用于模拟的α-Al2O3(0001)-水界面示意图。
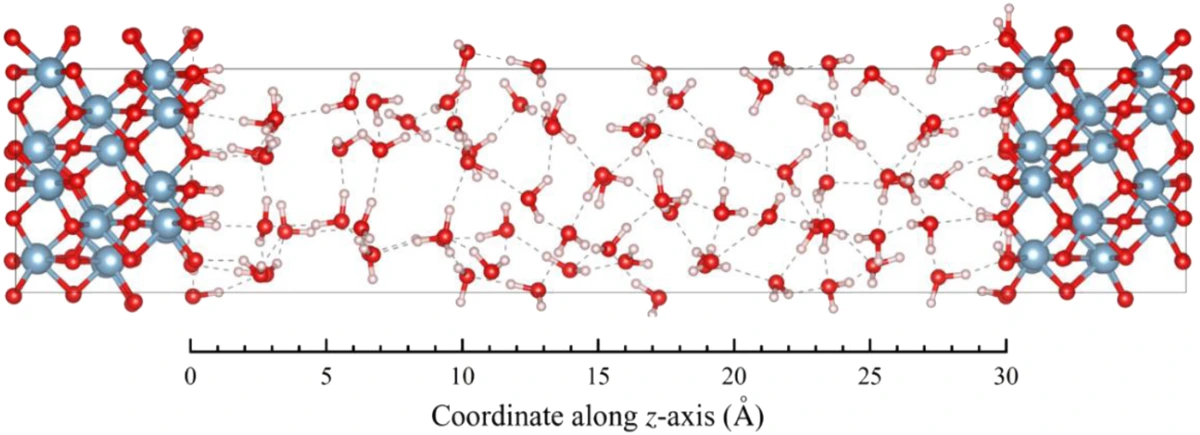
图2对比了基于从头算分子动力学模拟(AIMD)和机器学习加速的分子动力学模拟(DPMD)得到的轨迹结构,用于验证训练的DP模型的可靠性。结果表明AIMD和DPMD能够给出基本一样的结构信息。
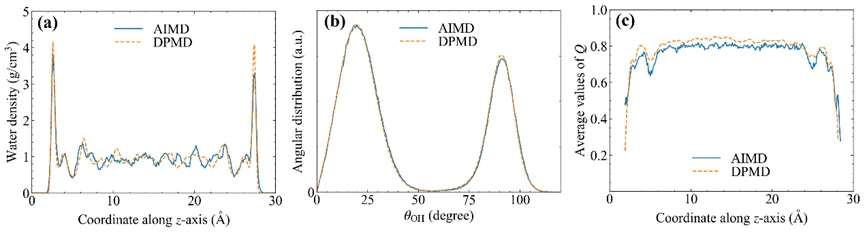
图3对比了基于DW模型计算得到的体相水的红外拉曼光谱和实验光谱,用于验证DW模型的可靠性。由于计算采用的PBE泛函给出的振动频率偏低,导致计算和实验光谱的振动频率有一定差距,但对应的光谱特征能够相互对应。
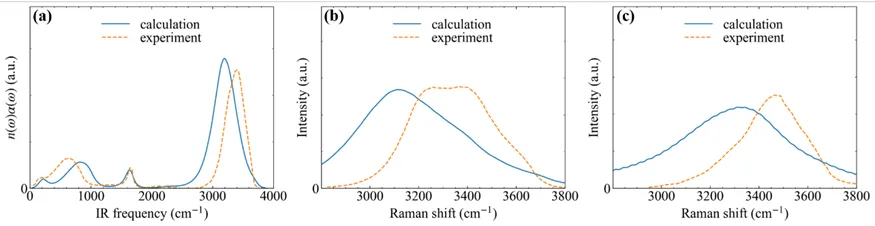
图4研究了基于偶极矩-极化率关联函数(μ-α)和表面敏感速度-速度关联函数(ssVVCF)两种方法计算 SFG 光谱的差异。结果表明,使用μ-α方法计算α-Al2O3(0001)-水界面的SFG光谱时,计算互关联项时需要考虑较大的截断半径。这是由于表面羟基和界面第一层水之间的相互作用对 SFG 光谱产生了显著影响。这种影响有别于不同截断半径对空气-水界面和疏水界面的 SFG 光谱的影响。
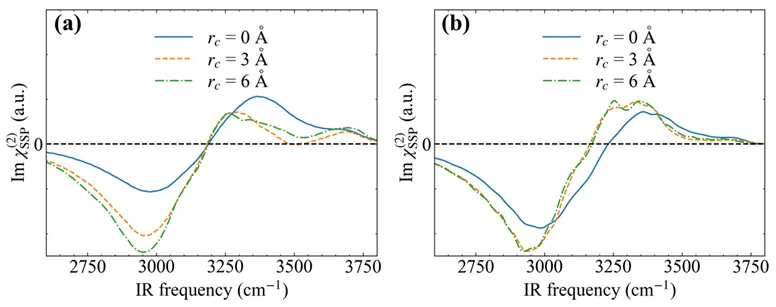
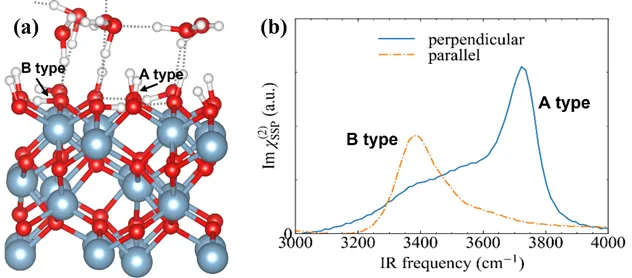
图5展示了两种不同类型的界面羟基和对应的SFG光谱,表明α-Al2O3(0001)表面存在着直立和水平的羟基,对应的振动频率也存在明显差异。直立羟基(A type)由于不能形成氢键有着较高的振动频率,而水平羟基(B type)由于和周围羟基的氧形成氢键振动频率较低。