Decoding the Competing Effects of Dynamic Solvation Structures on Nuclear Magnetic Resonance Chemical Shifts of Battery Electrolytes via Machine Learning
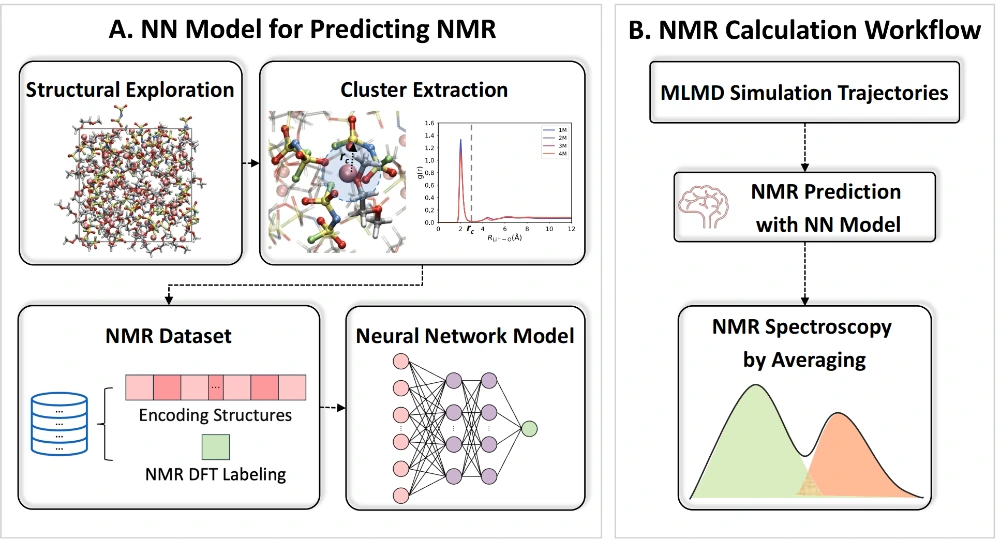
Understanding the solvation structure of electrolytes is critical for optimizing the electrochemical performance of rechargeable batteries as it directly influences properties such as ionic conductivity, viscosity, and electrochemical stability. The highly complex structures and strong interactions in high-concentration electrolytes make accurate modeling and interpretation of their "structure–property" relationships even more challenging with spectroscopic methods. In this study, we present a machine learning-based approach to predict dynamic 7Li NMR chemical shifts in LiFSI/DME electrolyte solutions. Additionally, we provide a comprehensive structural analysis to interpret the observed chemical shift behavior in experiments, particularly the abrupt changes in 7Li chemical shifts at high concentrations. Using advanced modeling techniques, we quantitatively establish the relationship between the molecular structure and NMR spectrum, offering critical insights into solvation structure assignments. Our findings reveal the coexistence of two competing local solvation structures that shift in dominance as electrolyte concentration approaches the concentrated limit, leading to an anomalous reversal of 7Li NMR chemical shift in the experiment. This work provides a detailed molecular-level understanding of the intricate solvation structures probed by NMR spectroscopy, leading the way for an enhanced electrolyte design.